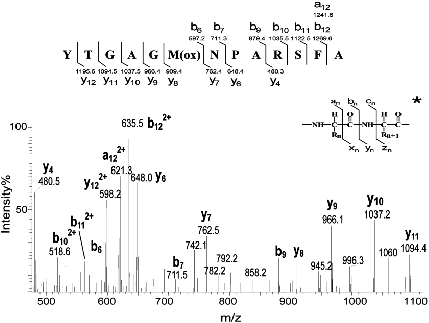
Figure 1: A typical spectrum for a peptide of m/z 680.0
De Novo Interpretation of Tandem Mass Spectrometry DataSubject proposed by Behshad Behzadibehzadi[at]lix.polytechnique.fr difficulté= moyen (**) |
Let us give evidences that how on can construct the sequence YTGAGMNPARSFA can be found from the given spectrum. The differences of the x coordinate of the three high consecutive peaks at 879.4, 1035.5 and 1122.5 (156.1 and 87.0) denote approximately the masses of the amino acids R and S respectively (see the url of the masses of amino acids presented in the next section). This shows that the substring RS is probably a substring (a tag) of the sequence. Different tags can be found in the similar way. Note that the tags can have different orientations. One way to find the best peptide sequence is to generate all the possible sequences and to choose the sequence which has the higest score. The score can for example be defined by the number of matched peaks or the length of the matched tags. This way of computation is too expensive in the terms of execution time. Dynamic programming and graph theoretical approaches have been propoed for computing efficiently a reliable sequence.
Figure 1: A typical spectrum for a peptide of m/z 680.0
This document was translated from LATEX by HEVEA.